Cisztationin-β-szintáz
| cystathionine beta-synthase | |
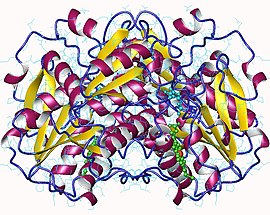 | |
| Cisztationin-β-szintáz-homodimer, humán | |
| Azonosítók | |
| Jel | CBS |
| Entrez | 875 |
| OMIM | 613381 |
| UniProt | P35520 |
| PDB | 1JBQ |
| Egyéb adatok | |
| EC-szám | 4.2.1.22 |
| Lokusz | 21. krom. q22.3 |
A cisztationin-β-szintáz, röviden CBS (EC 4.2.1.22) a CBS gén által kódolt enzim. A transzszulfurációs útvonal első lépését, a cisztationin homociszteinből való előállítását katalizálja:[1]
- l-Ser + l-Hcy ⇌ l-cisztationin + H2O
A CBS a piridoxál-foszfát (PLP) kofaktort használja, és allosztérikusan szabályozhatják effektorok, például az S-adenozil-l-metionin (adoMet). Ez az enzim liáz, egész pontosan C–O kötéseket bontó hidroliáz.
A CBS többdoménes enzim, részei egy N-terminális enzimatikus és két CBS domén. A CBS gén a homocisztinuriát okozó mutációk leggyakoribb lokusza.[2]
Nevezéktan
Az enzimosztály szabályos neve l-szerin-hidroliáz (homocisztein-hozzáadó, l-cisztationin-képző). További gyakori nevek:
- β-tionáz,
- cisztein-szintáz,
- l-szerin-hidroliáz (homocisztein-hozzáadó),
- metilcisztein-szintáz,
- szerin-szulfhidráz,
- szerin-szulfhidriláz.
A metilcisztein-szintáz EC-száma EC 4.2.1.23 lett 1961-ben. Ennek oka a CBS mellékreakciója. A 4.2.1.23 számot 1972-ben törölték.[3]
Szerkezet
A humán cisztationin-β-szintáz 551 aminosavas, 61 kDa-os alegységekből álló tetramer. Szerkezete 3 modulos, az N-terminális hemdomént a PLP kofaktort tartalmazó mag követi.[4] A kofaktor a hemdoménben van, Schiff-bázis köti meg.[5] A Schiff-bázis C=N kötést tartalmazó funkciós csoport, ahol a nitrogén aril- vagy alkilcsoporthoz kötődik. A hemdomén 70 aminosavas, feltehetően csak emlősök CBS-ében van jelen, élesztőkében- és protozoonokéban nem. A CBS C-terminális szabályzódoménjében két β-α-β-β-α CBS-domén ismétlődik, ez más fehérjékben is jelenlévő másodlagos szerkezeti jellemző.[4] A CBS C-terminális gátló domént tartalmaz. Ez intra- és allosztérikusan szabályozza az aktivitást, és a fehérje tetramer helyzetének fenntartásában fontos.[4] A gátlást az adoMet allosztérikus effektor vagy a szabályzó domén deléciója szüntetheti meg, azonban a hatások nagysága eltérhet.[4] Ezek mutációi összefüggnek örökletes betegségekkel.[6]
A hemdomén N-terminális kört tartalmaz, mely a hemet köti, és a C52 és H65 axiális ligandumokat adja. A hem PLP-kötőhelytől való távolsága alapján az nem játszik szerepet a katalízisben, azonban a hemdomén deléciója a redoxiszenzitivitás csökkenésével jár, így feltehetően a hem redoxiszenzor.[5] A protoporfirin IX jelenléte a CBS-ban egyedi PLP-dependens enzimmé teszi, és csak az emlős-CBS-ben található meg. A D. melanogaster és a D. discoides rövidebb N-terminális kiterjesztéssel rendelkezik, így a hisztidin és a cisztein akadályozva vanna. Azonban az Anopheles gambiae N-terminális kiterjesztése hosszabb a humán enziménél, és tartalmazza a hisztidint és a ciszteint. Így feltehetően a nyálkagomba- és rovar-CBS hemoprotein, így a hemdomén feltehetően korán jelent meg, az állatok és a nyálkagombák elválása előtt.[4] A PLP K119-et aktív helyén tartalmazó Schiff-bázist alkotó belső aldimin. A katalitikus és a szabályzó domének közt hiperérzékeny hely található, mely proteolitikus bontást okoz, és rövidebb, az eredetinél aktívabb dimer enzimet tartalmaz. Az élesztőben talált egyik enzimváltozatot se szabályozza az adoMet. Az élesztőenzimet a dimert adó C-terminális deléció is aktiválja.[4]
2007 végéig 2 harmadlagos szerkezete ismert a CBS enzimeknek, ezek kódja PDB: 1JBQ és PDB: 1M54.
Enzimaktivitás
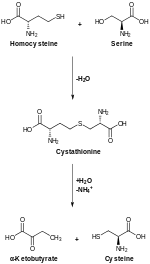
A CBS katalizálta transzszulfuráció a homociszteint cisztationinná alakítja, ezt a cisztationin-γ-liáz ciszteinné bontja.[7]
A CBS az emlősök kénmetabolizmusában fontos, mert a metioninmegtartás és a ciszteinné alakítás kérdését dönti el. Továbbá a transzszulfurációs útvonal az egyetlen felesleges kéntartalmú aminosavak eltávolítására képes út.[4]
Más β-helyettesítő enzimekhez hasonlóan a CBS katalizálta reakció feltehetően adoMet-kötött köztitermékeket tartalmaz. A szerin hozzáadása külső aldimint adó transzschiffizációs reakciót ad. Ez protont von el az α-szénatomnál, majd elmináció útján aminoakrilát köztiterméket ad. A homocisztein és a nukleofil homocisztein-tiolát reakciója az aminoakriláton és a Cα reprotonációja adják a cisztationin külső aldiminjét. Transzaldiminációval jön létre a l-cisztationin végtermék.[4] Ez aminoakrilát intermediert is alkothat, ez alapján a CBS reakciója visszafordítható.[8]
Az enzimkatalizált reakció mért V0-ja általában az állandó állapotnak felel meg, ahol [ES] konstans, bár ez a reakció korai részére korlátozódik, e kezdeti sebességek elemzése az állandó állapotú kinetika. Az élesztő-CBS állandó állapotú elemzése párhuzamos vonalakat ad. Ezen eredmények a feltételezett pingpongmechanizmusnak felelnek meg, ahol a szerinkötést és a vízfelszabadítást követik a homocisztein-kötés és a cisztationin-felszabadítás. Azonban a patkány-CBS enzimkinetikai elemzése egymást metsző vonalakat ad: a szerinen lévő β-szubsztituens a homocisztein kötése előtt nem válik le az enzimtől.[4]
Egy CBS-t tartalmazó alternatív reakció a cisztein homociszteinnel való, cisztationint és kén-hidrogént (H2S) adó kondenzációja.[8] A CBS az agyban H2S-t termel l-ciszteinből. Ez az út is adoMet-függő.[9]
A CBS enzimaktivitása nincs jelen minden szövetben és sejtben: a patkányszívben, -tüdőben, -herékben, -mellékvesékben és -lépben nem aktív. Az emberi szívizomban és humán aortaendotélsejt-kultúrákban inaktív. E szövetek CBS-hiánya alapján e szövetek nem képesek ciszteinszintézisre, így külső ciszteinforrást igényelnek. Továbbá e szöveteknek nagyobb lehet a homocisztein-szenzitivitásuk, mivel a homocisztein-többlet transzszulfurációs katabolizmusára nem képesek.[8]
Szabályzás
A CBS adoMet általi allosztérikus aktiválása határozza meg a homocisztein sorsát. Az emlős-CBS-t az adoMet 2,5-5-szörösen aktiválja 15 μM disszociációs állandó mellett.[2] Az adoMet allosztérikus aktivátor, mely növeli a CBS-reakció Vmax-át, de nem befolyásolja a szubsztrátok Km-ét. Vagyis az adoMet szubsztrátkötés helyett az enzimműködés gyorsításával növeli a CBS aktivitását.[4] A fehérje feltehetően az allosztérikus szabályzás morfiinmodelljét használja.[10]
A humán CBS fontos lépést hajt végre a cisztein-bioszintézisben az adoMet szabályzó irányítópontjának biztosításával. A homocisztein a metioninná metiláció után számos anyagot, például neurotranszmittereket, fehérjéket és nukleinsavakat metiláló adoMet-ná alakítható. Az adoMet saját bioszintézisét irányító allosztérikus CBS-aktivátor: az alacsony adoMet-koncentráció alacsony CBS-aktivitást okoz, a homocisztein az adoMet-szintézissel járó transzmetilációs ciklusba kerül, a magas adoMet-koncentráció a homociszteint a cisztein-bioszintézist okozó transzszulfurációs útvonalba irányítja.[11]
Az emlősökben a CBS erősen szabályzott enzim, redoxiszenzorként működő hemkofaktorral,[6] mely az aktivitását a redoxipotenciál változásainak megfelelően modulálhatja. Ha a nyugalomban lévő CBS Fe2+-hemet tartlmaz, lehetséges az enzim aktivációja oxidatív körülmények közt Fe3+-hemmé való oxidációval.[4] A Fe2+-enzim CO- vagy NO-kötéssel gátolható, az enzim aktivitása kétszer akkora a Fe2+ Fe3+-má való oxidációjával. A hem redoxiállapota pH-dependens: a Fe2+-CBS Fe3+-CBS-sé való oxidációja alacsony pH mellett kedvezőbb.[12]
Mivel míg az emlős CBS-ben van, az élesztők és a Trypanosoma cruzi CBS-ében nincs hemkofaktor, Ruma et al. szerint a hem nem szükséges a CBS-aktivitáshoz.[4]
A CBS-transzkripciót szabályozzák az NF-Y, az SP-1 és az SP-3. Erősítik a transzkripciót továbbá a glükokortikoidok és a glikogén, gyengíti az inzulin. A metionin a CBS-t poszttranszkripciósan erősíti.
Humán betegségekben
- A Down-szindrómára jellemző a cisztationin-β-szintáz túlexpressziója és a vér alacsony homociszteinszintje. Feltehetően a cisztationin-β-szintáz túlexpressziója a fő felelős a betegségért a GabaA és a Dyrk1a diszfunkciója mellett. A Down-szindróma fenotípusa az alább leírt hiperhomociszteinémiával ellentétes. CBS-gátló gyógyszereket szabadalmaztatott a Jérôme Lejeune Alapítvány 2011 novemberében, és állat- és emberkísérleteket terveznek.
- A hiperhomociszteinémia a vér különösen magas homociszteinszintjét jelenti. A CBS-mutációk az öröklött hiperhomociszteinémia leggyakoribb okai. Az MTHFR-, MTR- és MTRR/MS-útvonalakat érintő genetikai hibák is közrejátszhatnak a magas homociszteinszintekben. A veleszületett CBS-hibák hiperhomociszteinémiát okozhatnak, ahol a szív- és érrendszeri komplikációk korai agresszív artériabetegséget okozhatnak. A hiperhomociszteinémia 3 másik szervrendszert, a látórendszert, a központi idegrendszert és a vázrendszert is érintik.
- A CBS-elégtelenség okozta homocisztinuria speciális hiperhomociszteinémia-típus. Ritka, öröklött autoszomális recesszív, általában gyermekkorban diagnosztizált betegség. 131 homocisztinuriát okozó mutáció ismert. A CBS-domén-mutációk közös funkciós jellemzője az adoMet általi aktiváció megszűnte vagy nagymértékű csökkenése.[11] Nem ismert a homocisztinuriára specifikus gyógyszer, de sokakat kezelnek nagy mennyiségű B6-vitaminnal, a CBS kofaktorával.
Biológiai mérnökség
A CBS részt vesz a petesejtfejlődésben. Azonban kevés ismeret van a CBS regionális és sejtbeli expressziójáról a petefészekben, és a petefészekben lévő tüszőfejlődés alatti helye és expressziója ismeretlen.[13]
A CBS-hiány egerekben terméketlenséget okoz a méhbeli fehérjeexpresszió csökkenése miatt.[14]
Mutációk
A CBS-expressziót irányító gének nem feltétlenül működnek teljesen SNP (egypontos nukleotid-polimorfizmus) esetén. Ismert változatok például az A360A, a C699T, az I278T, az N212N és a T42N. Ezek, melyek a hatékonyságot befolyásolhatják, DNS-teszttel azonosíthatók.
Jegyzetek
- ↑ Entrez Gene: CBS cystathionine-beta-synthase
- ↑ a b Janosík M, Kery V, Gaustadnes M, Maclean KN, Kraus JP (2001. szeptember 1.). „Regulation of human cystathionine beta-synthase by S-adenosyl-L-methionine: evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region”. Biochemistry 40 (35), 10625–33. o. DOI:10.1021/bi010711p. PMID 11524006.
- ↑ EC 4.2.1.23
- ↑ a b c d e f g h i j k l Banerjee R, Zou CG (2005. január 1.). „Redox regulation and reaction mechanism of human cystathionine-beta-synthase: a PLP-dependent hemesensor protein”. Archives of Biochemistry and Biophysics 433 (1), 144–156. o. DOI:10.1016/j.abb.2004.08.037. PMID 15581573.
- ↑ a b Yamanishi M, Kabil O, Sen S, Banerjee R (2006. december 1.). „Structural insights into pathogenic mutations in heme-dependent cystathionine-beta-synthase”. Journal of Inorganic Biochemistry 100 (12), 1988–95. o. DOI:10.1016/j.jinorgbio.2006.08.020. PMID 17069888.
- ↑ a b Kabil O, Zhou Y, Banerjee R (2006. november 1.). „Human cystathionine beta-synthase is a target for sumoylation”. Biochemistry 45 (45), 13528–36. o. DOI:10.1021/bi0615644. PMID 17087506.
- ↑ Nozaki T, Shigeta Y, Saito-Nakano Y, Imada M, Kruger WD (2001. március 1.). „Characterization of transsulfuration and cysteine biosynthetic pathways in the protozoan hemoflagellate, Trypanosoma cruzi. Isolation and molecular characterization of cystathionine beta-synthase and serine acetyltransferase from Trypanosoma”. The Journal of Biological Chemistry 276 (9), 6516–23. o. DOI:10.1074/jbc.M009774200. PMID 11106665.
- ↑ a b c Jhee KH, Kruger WD (2005). „The role of cystathionine beta-synthase in homocysteine metabolism”. Antioxidants & Redox Signaling 7 (5–6), 813–22. o. DOI:10.1089/ars.2005.7.813. PMID 15890029.
- ↑ Eto K, Kimura H (2002. november 1.). „A novel enhancing mechanism for hydrogen sulfide-producing activity of cystathionine beta-synthase”. The Journal of Biological Chemistry 277 (45), 42680–5. o. DOI:10.1074/jbc.M205835200. PMID 12213817.
- ↑ T. Selwood (2011). „Dynamic dissociating homo-oligomers and the control of protein function”. Arch. Biochem. Biophys. 519 (2), 131–143. o. DOI:10.1016/j.abb.2011.11.020. PMID 22182754.
- ↑ a b Ignoul S, Eggermont J (2005. december 1.). „CBS domains: structure, function, and pathology in human proteins”. American Journal of Physiology. Cell Physiology 289 (6), C1369–78. o. DOI:10.1152/ajpcell.00282.2005. PMID 16275737.
- ↑ Puranik M, Weeks CL, Lahaye D, Kabil O, Taoka S, Nielsen SB, Groves JT, Banerjee R, Spiro TG (2006. május 1.). „Dynamics of carbon monoxide binding to cystathionine beta-synthase”. The Journal of Biological Chemistry 281 (19), 13433–8. o. DOI:10.1074/jbc.M600246200. PMID 16505479.
- ↑ Liang R, Yu WD, Du JB, Yang LJ, Shang M, Guo JZ (2006. november 1.). „Localization of cystathionine beta synthase in mice ovaries and its expression profile during follicular development”. Chinese Medical Journal 119 (22), 1877–83. o. DOI:10.1097/00029330-200611020-00006. PMID 17134586.
- ↑ Guzmán MA, Navarro MA, Carnicer R, Sarría AJ, Acín S, Arnal C, Muniesa P, Surra JC, Arbonés-Mainar JM, Maeda N, Osada J (2006. november 1.). „Cystathionine beta-synthase is essential for female reproductive function”. Human Molecular Genetics 15 (21), 3168–76. o. DOI:10.1093/hmg/ddl393. PMID 16984962.
Fordítás
- Ez a szócikk részben vagy egészben a Cystathionine beta synthase című angol Wikipédia-szócikk ezen változatának fordításán alapul. Az eredeti cikk szerkesztőit annak laptörténete sorolja fel. Ez a jelzés csupán a megfogalmazás eredetét és a szerzői jogokat jelzi, nem szolgál a cikkben szereplő információk forrásmegjelöléseként.
Források
- Kraus JP (1994). „Komrower Lecture. Molecular basis of phenotype expression in homocystinuria”. J. Inherit. Metab. Dis. 17 (4), 383–90. o. DOI:10.1007/BF00711354. PMID 7967489.
- Kraus JP, Janosík M, Kozich V (1999). „Cystathionine beta-synthase mutations in homocystinuria”. Hum. Mutat. 13 (5), 362–75. o. DOI:<362::AID-HUMU4>3.0.CO;2-K 10.1002/(SICI)1098-1004(1999)13:5<362::AID-HUMU4>3.0.CO;2-K. PMID 10338090.
- Jones AL (1999). „The localization and interactions of huntingtin”. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354 (1386), 1021–7. o. DOI:10.1098/rstb.1999.0454. PMID 10434301.
- Griffiths R, Tudball N (1977). „The molecular defect in a case of (cystathionine beta-synthase)-deficient homocystinuria”. Eur. J. Biochem. 74 (2), 269–73. o. DOI:10.1111/j.1432-1033.1977.tb11390.x. PMID 404147.
- Kraus J, Packman S, Fowler B, Rosenberg LE (1978). „Purification and properties of cystathionine beta-synthase from human liver. Evidence for identical subunits”. J. Biol. Chem. 253 (18), 6523–8. o. DOI:10.1016/S0021-9258(19)46963-9. PMID 681363.
- Longhi RC, Fleisher LD, Tallan HH, Gaull GE (1977). „Cystathionine beta-synthase deficiency: a qualitative abnormality of the deficient enzyme modified by vitamin B6 therapy”. Pediatr. Res. 11 (2), 100–3. o. DOI:10.1203/00006450-197702000-00003. PMID 840498.
- Kozich V, Kraus JP (1993). „Screening for mutations by expressing patient cDNA segments in E. coli: homocystinuria due to cystathionine beta-synthase deficiency”. Hum. Mutat. 1 (2), 113–23. o. DOI:10.1002/humu.1380010206. PMID 1301198.
- Münke M, Kraus JP, Ohura T, Francke U (1988). „The gene for cystathionine beta-synthase (CBS) maps to the subtelomeric region on human chromosome 21q and to proximal mouse chromosome 17”. Am. J. Hum. Genet. 42 (4), 550–9. o. PMID 2894761.
- Hu FL, Gu Z, Kozich V (1994). „Molecular basis of cystathionine beta-synthase deficiency in pyridoxine responsive and nonresponsive homocystinuria”. Hum. Mol. Genet. 2 (11), 1857–60. o. DOI:10.1093/hmg/2.11.1857. PMID 7506602.
- Sperandeo MP, Panico M, Pepe A (1995). „Molecular analysis of patients affected by homocystinuria due to cystathionine beta-synthase deficiency: report of a new mutation in exon 8 and a deletion in intron 11”. J. Inherit. Metab. Dis. 18 (2), 211–4. o. DOI:10.1007/BF00711769. PMID 7564249.
- Chassé JF, Paly E, Paris D (1995). „Genomic organization of the human cystathionine beta-synthase gene: evidence for various cDNAs”. Biochem. Biophys. Res. Commun. 211 (3), 826–32. o. DOI:10.1006/bbrc.1995.1886. PMID 7598711.
- Shih VE, Fringer JM, Mandell R (1995). „A missense mutation (I278T) in the cystathionine beta-synthase gene prevalent in pyridoxine-responsive homocystinuria and associated with mild clinical phenotype”. Am. J. Hum. Genet. 57 (1), 34–9. o. PMID 7611293.
- Kluijtmans LA, Blom HJ, Boers GH (1995). „Two novel missense mutations in the cystathionine beta-synthase gene in homocystinuric patients”. Hum. Genet. 96 (2), 249–50. o. DOI:10.1007/BF00207394. PMID 7635485.
- Sebastio G, Sperandeo MP, Panico M (1995). „The molecular basis of homocystinuria due to cystathionine beta-synthase deficiency in Italian families, and report of four novel mutations”. Am. J. Hum. Genet. 56 (6), 1324–33. o. PMID 7762555.
- Marble M, Geraghty MT, de Franchis R (1995). „Characterization of a cystathionine beta-synthase allele with three mutations in cis in a patient with B6 nonresponsive homocystinuria”. Hum. Mol. Genet. 3 (10), 1883–6. o. DOI:10.1093/hmg/3.10.1883. PMID 7849717.
- Kraus JP, Le K, Swaroop M (1994). „Human cystathionine beta-synthase cDNA: sequence, alternative splicing and expression in cultured cells”. Hum. Mol. Genet. 2 (10), 1633–8. o. DOI:10.1093/hmg/2.10.1633. PMID 7903580.
- de Franchis R, Kozich V, McInnes RR, Kraus JP (1995). „Identical genotypes in siblings with different homocystinuric phenotypes: identification of three mutations in cystathionine beta-synthase using an improved bacterial expression system”. Hum. Mol. Genet. 3 (7), 1103–8. o. DOI:10.1093/hmg/3.7.1103. PMID 7981678.
- Kruger WD, Cox DR (1994). „A yeast system for expression of human cystathionine beta-synthase: structural and functional conservation of the human and yeast genes”. Proc. Natl. Acad. Sci. U.S.A. 91 (14), 6614–8. o. DOI:10.1073/pnas.91.14.6614. PMID 8022826.
- Kozich V, de Franchis R, Kraus JP (1993). „Molecular defect in a patient with pyridoxine-responsive homocystinuria”. Hum. Mol. Genet. 2 (6), 815–6. o. DOI:10.1093/hmg/2.6.815. PMID 8353501.
További információk
- CBS Main Page at University of Colorado Health Sciences Center
- Cystathionine beta-synthase in BRENDA: The Comprehensive Enzyme Information System[halott link]
- Cystathionine beta-Synthase: Protein Data Bank Entry







